









合作伙伴:
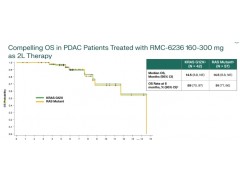
胰腺癌
抗癌药品百科
全球肿瘤医生网不具备药品销售资格,不售卖任何药物,也不帮助患者代购药物。网上药品代购存在假药等风险,且代购均为违法行为,广大患者做好自我保护。
")
Sunitinib(舒尼替尼)
适应症:...
- 药物介绍
- 药物靶点
- 相关资讯
- 临床试验
Sunitinib(舒尼替尼)药物介绍
| 【通用名称】 | 舒尼替尼 |
| 【药品名称】 | votrient |
| 【英文名称】 | Pazopanib |
| 【汉语拼音名称】 | pazuopani |
| 【美国上市时间】 | |
| 【靶点】 | RTK |
| 【主要成分】 | (Z)-N-[2-(二乙胺基)乙基-5-[(5-氟-2-氧代-1,2-二氢-3H-吲哚-3-亚基)甲基]-2,4-二甲基-3-氨甲酰-1H-吡咯苹果酸盐 |
| 【药品性状】 | 本品为胶囊剂,内容物为黄色至橙色的颗粒。1)瓶装 包装规格:14粒/瓶;28粒/瓶;30粒/瓶 2)铝箔/PVC/Aclar水泡眼包装 包装规格:28粒/盒 |
| 【用法用量】 | 本品治疗胃肠间质瘤和晚期肾细胞癌的推荐剂量是50mg,每日一次,口服,服药4周,停药2周(4/2给药方案)。 对于胰腺神经内分泌瘤,本品推荐剂量为37.5mg,口服,每日一次,连续服药,无停药期。 与食物同服或不同服均可。 |
| 【适应症】 | 1)甲磺酸伊马替尼治疗失败或不能耐受的胃肠间质瘤(GIST)。 2)不能手术的晚期肾细胞癌(RCC)。 3) 不可切除的,转移性高分化进展期胰腺神经内分泌瘤(pNET)成年患者。 |
| 【注意事项】 | 1)观察到增加血清转氨酶水平和胆红素。曾发生严重致死性肝毒性。开始治疗前和治疗期间定期测定肝化学。 2)曾观察到QT间隔延长和尖端扭转型室速(torsades de pointes)。较高危发生QT间隔延长患者慎用。 应考虑监查心电图和电解质。 3) 曾报道致死性出血事件。尚未在既往6个月内有咯血、脑、或有临床意义胃肠道出血史的患者研究VOTRIENT而不应在这些患者中使用。 4) 曾观察到动脉血栓形成事件和可能致死。对这些事件风险增加患者中慎用。 5) 曾发生胃肠道穿孔或瘘管。曾发生致死性穿孔事件。胃肠道穿孔或瘘管风险增加患者中慎用。 6) 曾观察到高血压。开始用VOTRIENT应充分控制血压。需要时监查和治疗高血压。 7) 在进行手术患者中建议中断VOTRIENT治疗。 8) 可能发生甲状腺机能减退。建议监查甲状腺功能。 9)蛋白尿:监查尿蛋白。对4级蛋白尿中断药物。 10) 当妊娠妇女给予VOTRIENT可能危害胎儿。怀孕潜能妇女应忠告对胎儿的潜在危害和服用是避免受孕。 |
| 【不良反应】 | 不良反应(≥20%)是疲劳、乏力、发热、腹泻、恶心、粘膜炎/口腔炎、呕吐、消化不良、腹痛、便秘、高血压、外周水肿、皮疹、手足综合征、皮肤褪色、皮肤干燥、毛发颜色改变、味觉改变、头痛、背痛、关节疼痛、肢端疼痛、咳嗽、呼吸困难、厌食和出血。 |
| 【禁忌】 | 无. |
| 【药理毒性】 | 苹果酸舒尼替尼是一种能抑制多个受体酪氨酸激酶(RTK)的小分子,其中某些受体酪氨酸激酶参与肿瘤生长、病理性血管形成和肿瘤转移的过程。通过对舒尼替尼抑制各种激酶(80多种激酶)的活性进行评价,证明舒尼替尼可抑制血小板衍生生长因子受体(PDGFRα和PDGFRβ)、血管内皮生长因子受体(VEGFR1、VEGFR2和VEGFR3)、干细胞因子受体(KIT)、Fms样酪氨酸激酶-3(FLT3)、1型集落刺激因子受体(CSF-1R)和神经胶质细胞系衍生的神经营养因子受体(RET)。生化和细胞测定证实舒尼替尼能抑制这些受体酪氨酸激酶(RTK)的活性,并在细胞增殖测定中证明了舒尼替尼的抑制作用。生化和细胞测定表明主要代谢物与舒尼替尼活性相似。 |
| 【药物相互作用】 |
苹果酸舒尼替尼是一种能抑制多个受体酪氨酸激酶(RTK)的小分子,其中某些受体酪氨酸激酶参与肿瘤生长、病理性血管形成和肿瘤转移的过程。通过对舒尼替尼抑制各种激酶(80多种激酶)的活性进行评价,证明舒尼替尼可抑制血小板衍生生长因子受体(PDGFRα和PDGFRβ)、血管内皮生长因子受体(VEGFR1、VEGFR2和VEGFR3)、干细胞因子受体(KIT)、Fms样酪氨酸激酶-3(FLT3)、1型集落刺激因子受体(CSF-1R)和神经胶质细胞系衍生的神经营养因子受体(RET)。生化和细胞测定证实舒尼替尼能抑制这些受体酪氨酸激酶(RTK)的活性,并在细胞增殖测定中证明了舒尼替尼的抑制作用。生化和细胞测定表明主要代谢物与舒尼替尼活性相似。 在表达受体酪氨酸激酶靶点的肿瘤模型的体内试验中,舒尼替尼能抑制多个受体酪氨酸激酶(PDGFRβ、VEGFR2、KIT)的磷酸化进程;在某些动物肿瘤模型中显示出抑制肿瘤生长或导致肿瘤消退,和/或抑制肿瘤转移的作用。体外实验结果表明苹果酸舒尼替尼能抑制靶向受体酪氨酸激酶(PDGFR、RET或KIT)表达失调的肿瘤细胞生长,体内试验结果表明其能抑制PDGFRβ-和VEGFR2-依赖的肿瘤血管形成。 毒理研究 重复给药毒性:一项3个月的猴重复给药试验(2,6,12mg/kg/日剂量)研究了药物对雌性生殖系统的影响,12 mg/kg/日剂量(约为推荐人用日剂量[RDD]的AUC的5.1倍)观察到卵巢变化(卵泡发育下降);在≥2 mg/kg/日剂量(约为推荐人用日剂量[RDD]的AUC的0.4倍)观察到子宫变化(子宫内膜萎缩)。在一项9个月的猴重复给药研究中,6mg/kg/日剂量时除阴道萎缩外,还对子宫和卵巢有影响(每日给药0.3,1.5和6mg/kg/日剂量,连续给药28天,停药14天,6 mg/kg/日剂量产生的平均AUC约为推荐人用日剂量[RDD] AUC的0.8倍)。3个月的研究中未明确无毒性效应水平;9个月的研究中无毒性效应剂量水平为1.5mg/kg/日。 生殖毒性:虽然舒尼替尼不影响大鼠的生育能力,但可能损害人的生育能力。雌性大鼠每天给药≤5.0mg/kg/日剂量未发现生殖毒性(0.5,1.5,5.0mg/kg/日剂量,给药21天直至妊娠第7天,5mg/kg/日剂量产生的平均AUC约是推荐人用日剂量[RDD]AUC的5倍),然而在5.0 mg/kg 剂量水平观察到明显的胚胎致死性毒性。雄性大鼠在与未用药的雌性大鼠交配前,接受了58天剂量为1、3或10mg/kg/日的舒尼替尼,未发现生殖毒性。舒尼替尼剂量≤10mg/kg/日时,对生育能力、交配、受孕指数和精子检查(形态、精子数和活动度)都没有影响(10 mg/kg剂量产生的平均AUC约为推荐人用日剂量[RDD]50mg/日的AUC的25.8倍)。 遗传毒性和致癌性:在两种动物中进行了评估舒尼替尼致癌作用:rasH2转基因小鼠和斯普拉格-杜勒大鼠。这两种动物产生了相似的阳性结果。对舒尼替尼进行了系列的体外遗传毒性试验(细菌突变[Ames试验]、人淋巴细胞染色体畸变)和一项大鼠体内骨髓微核试验,舒尼替尼未引起遗传损害。尽管未对舒尼替尼的致癌作用进行研究,但给予rasH2转基因鼠舒尼替尼0,10,25,75或200mg/kg/日,连续28天或6个月时,≥25mg/kg的剂量下观察到胃十二指肠癌,血管肉瘤发病率增加,和/或胃粘膜增生。在rasH2转基因小鼠中进行每天8mg/kg的剂量用药后(不低于人推荐日剂量[RDD]AUC的0.7倍)未观察到增生性改变。转基因小鼠rasH2中使用舒尼替尼1和6个月治疗后所观察到的致癌作用与人体的关系尚不清楚。类似地,在一项为期2年的大鼠致癌研究中,以28天为周期给药,随后是7天的停药期,依此重复,结果在剂量低至1mg/kg/日(大约为患者接受推荐日剂量50 mg/日时AUC的0.9倍)时发现十二指肠癌。3mg/kg/日(大约为患者接受推荐日剂量50 mg/日时AUC的7.8倍)的高剂量时,发现十二指肠肿瘤发病率增加,并伴有胃粘膜细胞增生以及嗜铬细胞瘤和肾上腺增生发病率增加。对舒尼替尼进行了系列的体外遗传毒性试验(细菌突变[艾姆斯氏试验]、人淋巴细胞染色体畸变)和一项大鼠体内骨髓微核试验,舒尼替尼未引起遗传损害。 药代动力学 尚缺乏中国人群的药代动力学研究数据,以下均为来自国外的人体药代动力学研究数据。 已在135例健康志愿者和266例实体瘤受试者中评价了舒尼替尼和苹果酸舒尼替尼的药代动力学。 吸收、分布、代谢和排泄 一般在口服给药后6~12小时(Tmax)舒尼替尼达到最大血浆浓度(Cmax)。进食对舒尼替尼生物利用度无影响。与食物同服或不同服均可。 体外实验表明舒尼替尼及其主要活性代谢物的人血浆蛋白结合率分别为95%和90%,在100~4000 ng/ml 范围内无浓度依赖。舒尼替尼的表观分布容积 (Vd/F) 为 2230L。在25~100mg的剂量范围内, 血浆药时曲线下面积(AUC)和最大血浆浓度(Cmax)随剂量成比例增加。 舒尼替尼主要由细胞色素P450 CYP3A4代谢,产生的主要活性代谢物被CYP3A4进一步代谢。其主要活性代谢物占总暴露量的23~37%。主要通过粪便排泄。在一项[C]标记的舒尼替尼质量平衡的人体试验中,剂量的61%是通过粪便排出,而肾脏排泄的药物和代谢物约占剂量的16%。舒尼替尼和主要活性代谢物在血浆、尿和粪便中发现的主要药物相关成分,分别代表了合并标本中的91.5 %、86.4 %和73.8%的放射活性。尿和粪便中能检测到次要代谢物,但在血浆中一般未能发现。总口服清除率(CL/F)为34~62升/小时,受试者间的变异系数为40%。 健康志愿者口服单剂量舒尼替尼后,舒尼替尼和主要活性代谢物的终末半衰期分别为40~60小时和80~110小时。每日重复给药后,舒尼替尼蓄积3~4倍,而其主要代谢物蓄积7~10倍,在10~14天内舒尼替尼和主要活性代谢物达稳态浓度。第14天血浆舒尼替尼和主要活性代谢物的总浓度为62.9~101 ng/ml。每日重复给药或按治疗方案重复周期给药,未发现舒尼替尼和主要活性代谢物的药代动力学有明显的变化。 受试的健康志愿者和实体瘤受试者的药代动力学相似,包括胃肠间质瘤(GIST)和晚期/转移性肾细胞癌(RCC)受试者。 特殊群体 群体药代动力学分析的人口学数据表明年龄、体重、肌酐清除率、人种、性别或ECOG体力状态评分对舒尼替尼或其活性代谢物的药代动力学没有临床相关性影响。 未进行舒尼替尼在儿童患者中的药代动力学评价。 肝功能不全 Child-Pugh A级或Child-Pugh B级肝功能损害的患者接受舒尼替尼治疗无需调整初始剂量。舒尼替尼及其主要代谢产物主要由肝脏代谢。与肝功能正常的受试者相比,单剂舒尼替尼在轻度 (Child-Pugh A级) 或中度(Child-Pugh B级)肝功能损害的受试者中系统暴露量是相似的。未在重度(Child-Pugh C级)肝功能损害患者进行研究。在癌症患者中进行的临床研究排除了ALT或AST >2.5倍ULN,或因肝转移ALT或AST >5.0倍ULN的患者。 肾功能不全 与肾功能正常(CLcr>80 ml/分钟)的受试者相比,单剂舒尼替尼在重度肾功能损害(CLcr<30 ml/分钟)的受试者中系统暴露量是相似的。 轻度、中度及重度肾功能损害的患者接受舒尼替尼治疗无需调整初始剂量。后续剂量调整应基于患者安全性及耐受性见【剂量调整】。血液透析的末期肾病患者(ESRD)无需调整初始剂量。舒尼替尼在血液透析的末期肾病患者中暴露量比肾功能正常的患者低47%。因此,后续剂量可能需根据患者的安全性和耐受性逐步比初始剂量增加一倍。
|
| 【孕妇及哺乳用药】 | 孕妇接受舒尼替尼治疗可能会伤害胎儿。由于血管形成是胚胎和胎儿发育的关键,舒尼替尼抑制血管形成,可能对妊娠产生不良作用。 |
| 【贮藏】 | 保存于25℃;允许范围为15~30℃ |
| 【有效期】 | 36个月 |
| 【批准文号】 | JX20060174 进口药品注册证号 (1) 12.5mg: H20130258;H20130275 (2) 25mg: H20130259;H20130276 (3) 37.5mg:H20130260;H20130277 (4) 50mg: H20130261;H20130278 |
Sunitinib(舒尼替尼)药物靶点
Sunitinib(舒尼替尼)药物相关资讯
Sunitinib(舒尼替尼)药物临床试验
Sunitinib(舒尼替尼)药物热门问答
Sunitinib(舒尼替尼)药物资讯
-

6款第一、二、三代铂类化疗药物顺铂、卡铂、环硫铂、奈达铂、奥沙利铂、洛铂大盘点
以临床最常用的化疗药物类型之一,铂类化疗药为例,铂类化疗药共分为三代,这些药物的适应症及不良反应之间有非常独特的区别,不同药物之间交叉耐药情况也有差异。
-

刚刚!各类癌症的五年生存率及化疗成功率统计数据出炉
关于化疗,各种流言从未间断:化疗杀敌一千,自损八百癌症患者是死于化疗,而不是癌症本身化疗副作用极其痛苦且让人死得更快....
-

化疗各种毒性反应 1~4 级是啥?有这几张表就够了
化疗不仅存在诸多不良反应,给人身带来不同程度的损伤,而且随着化疗疗程的加长,化疗的敏感性越来越差,甚至产生耐药,治疗效果
-

人参皂苷抗癌抗肿瘤,人参皂苷的作用和功效是可以联合放化疗、靶向药可减轻副作用,抑制癌细胞
参的主要药理活性成分人参皂苷已被证明具有多种药用效果,包括突出的抗癌活性。
-
癌症肿瘤患者怎么提高免疫力,胸腺肽的功能作用与功效可能90%的患者都不知道
人们可能因多种原因而免疫功能低下,包括高龄、代谢紊乱(如糖尿病)、癌症治疗,甚至癌症本身。
-

基因检测无突变怎么办,基因检测没有突变怎么办,别慌,化疗药也有全新进展
如果基因检测发现了突变自然是好的,意味着患者可以开始尝试各类靶向药物;但并非每位患者都能通过基因检测发现有药物的靶点,而且还有一部分患者,因为耐药等原因而陷入了无靶向药可用的境地。
-

收藏|如何对抗化疗期间的各种副作用?方法都总结在这了!
在癌症治疗领域,虽然靶向药和免疫疗法越来越普遍,但化疗、放疗还是很多癌症的首选治疗方案。由放化疗引起的副作用让很多患者的
-

【抗癌日报】-大麻联合化疗比单独化疗延长3倍生存期
1、研究发现能阻止乳腺癌转移的方法约翰斯霍普金斯大学的研究人员报告说,在实验室培养的小鼠组织中证实,乳房乳管周围的细胞层

















