


常见的抗体药物靶点和CART靶点有哪些
常见CAR-T药物与抗体药物的靶点
1、CD19
CD19是CAR-T细胞治疗白血病和淋巴瘤等血液系统恶性肿瘤临床试验中最常用的生物标志物。全球已完成和正在进行的CAR-T临床试验中,以CD19为靶点的CAR-T临床试验占了总数的50%。很显然CD19是目前CAR-T细胞治疗最重要的靶标分子。
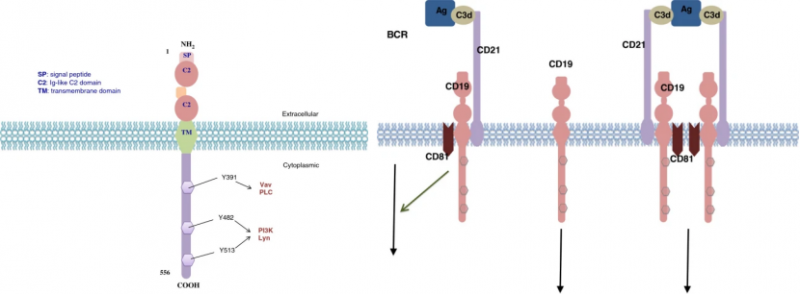
人CD19抗原是一种95kd的跨膜糖蛋白,属于免疫球蛋白(Ig)超家族。它由位于16号染色体短臂16p11.2上7.41kb的cd19基因编码。该基因包含15个外显子并编码含有556个氨基酸的CD19分子。该基因存在着不止一种的mRNA转录物,但在体内仅分离出两种转录物异构体。
CD19作为成熟B细胞表面多分子复合物的主要信号成分,可以与补体受体(CD21)、四跨膜蛋白(CD81)以及CD225在内的几种膜蛋白形成复合物,该类复合物降低了由抗原引发的B细胞活化的阈值。CD19是BCR信号转导的关键共同受体,BCR介导的信号传导需要蛋白酪氨酸激酶(PTK)激活,CD19则会募集并放大Src家族蛋白酪氨酸激酶的激活,例如Lyn和Fyn。在BCR激活后,CD19还通过募集和激活PI3K和下游Akt激酶来增强BCR诱导的对B细胞扩增至关重要的信号传导。
总的来说,CD19被认为在B细胞激活中发挥双重作用。首先,它作为一种衔接蛋白发挥作用,将细胞质信号蛋白募集到膜上;其次,CD19作为CD19/CD21复合物的信号亚基,可以增强抗原-补体-BCR-CD19/CD21所引起的B细胞活化。CD19虽然与B细胞的信号转导、增殖相关,但是目前并不知道CD19是否能够直接导致B细胞癌变,不过CD19在大多数B细胞肿瘤中都有表达,例如急性淋巴细胞白血病(ALL)、慢性淋巴细胞白血病(CLL)和B细胞淋巴瘤。
CD19已被一些抗体药物视为淋巴瘤和白血病治疗的重要治疗靶点。新的抗体类药物,如双特异性抗体、ADC也被设计用于靶向CD19。目前已上市的药物是MorphoSys的Tafasitamab,其他进展较快的药物包括安进的双抗Blinatumomab、赛诺菲的ADC药物SAR3419等。
以CD19为靶点的CAR-T药物则较多,例如KYMRIAH、YESCARTA、TECARTUS和BREYANZI等。
2、BCMA
B细胞成熟抗原(BCMA)或CD269,也被称为肿瘤坏死因子受体超家族成员17(TNFRSF-17),它在正常和恶性浆细胞(PC)中有高水平的表达。BCMA由位于16号染色体短臂(16p13.13)上的2.92-kb TNFRSF17基因编码,由被2个内含子隔开的3个外显子组成。BCMA是一个含有184个氨基酸20.2-kDa的III型跨膜糖蛋白,其细胞外N末端含有6个半胱氨酸的保守基序。
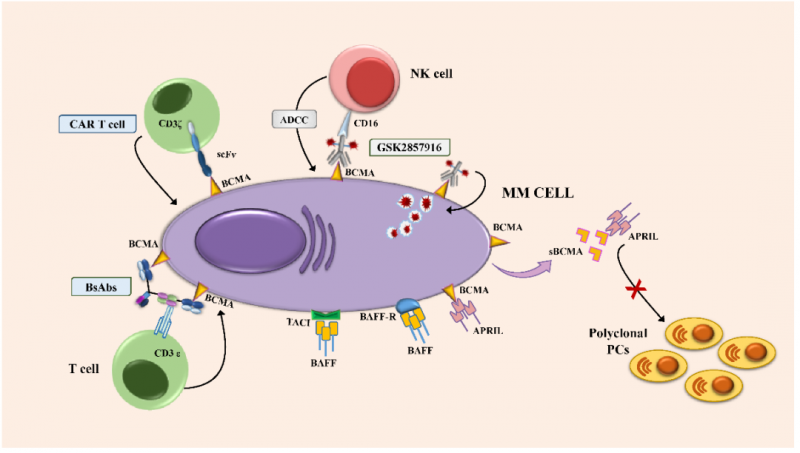
BCMA,连同其他两个功能相关的TNFR超家族成员,B细胞激活因子(BAFF,也称为BLyS受体,BAFF-R)和跨膜激活剂、钙调节剂、亲环蛋白配体相互作用物(TACI),协同调节B细胞的增殖成熟和存活,以及向浆细胞(PC)的分化。目前来说,针对BCMA靶点的抗体药物都还没有上市,例如葛兰素的ADC药物Belantamab mafodotin、安进的双抗AMG701都还处于临床试验阶段。但针对BCMA靶点的细胞治疗药物则有着更快的推进速度,例如BMS/Bluebird的ABECMA以及传奇生物的CARVYKTI都已经获得了上市资格。
3、HER2
HER2即人类表皮生长因子受体2,它们参与正常的细胞生长,但在一些癌细胞表面,例如乳腺癌等,HER2则往往会过度表达。HER2是一种分子量为185kDa 的跨膜蛋白,由位于17号染色体长臂上的(17q12–21.32)HER2(也称为erb-b2 receptor tyrosine kinase 2 [ERBB2]) 基因表达。HER2通常在乳腺和皮肤等多个器官以及胃肠道、呼吸道、生殖道和泌尿道中上皮细胞的细胞膜上表达。
在正常的乳腺上皮细胞中,HER2的表达水平较低(每个基因组有两个HER2基因拷贝;每个细胞表面表达20,000个HER2受体),而在HER2阳性的乳腺癌细胞中,HER2则以较高的水平表达着(基因组最多有25–50个HER2基因拷贝;每个细胞表面表达2,000,000个HER2受体)。除乳腺癌外,在其他类型的肿瘤中(包括胃癌、卵巢癌、结肠癌、膀胱癌、肺癌、子宫颈癌、头颈癌、食道癌以及子宫浆液性子宫内膜癌等)也有HER2过表达的报道。
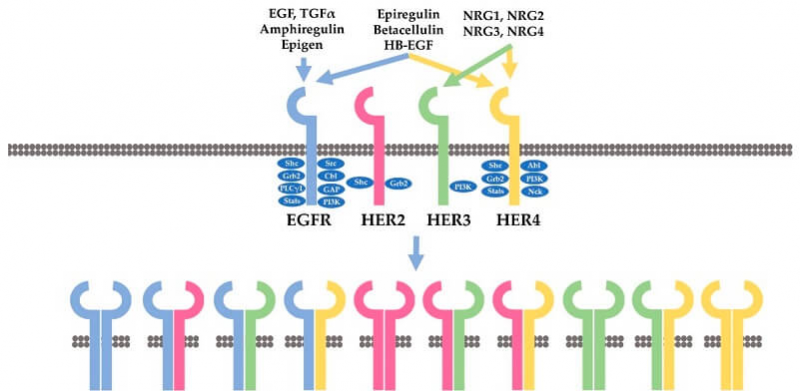
HER2属于表皮生长因子受体(EGFR)家族。该家族由四种HER受体组成:人表皮生长因子受体1(HER1)(也称为EGFR)、HER2、HER3和HER4。HER家族成员是跨膜受体酪氨酸激酶。它们既是受体,又是酶,能够同配体结合,并将细胞内靶蛋白上的酪氨酸残基磷酸化,即将三磷酸腺苷(ATP)的γ磷酸转移到蛋白质底物的酪氨酸残基上。
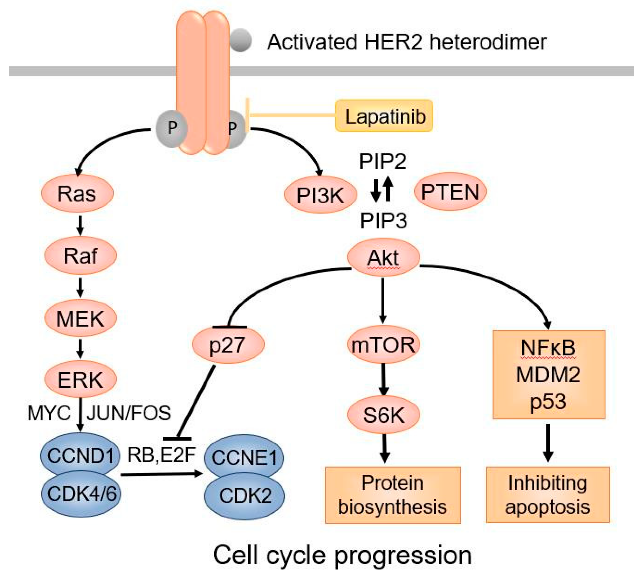
HER受体激活后,特定酪氨酸残基被磷酸化、随后的下游信号蛋白被募集和激活从而激活下游信号通路,进而促进细胞的增殖、存活、迁移、粘附、分化和血管生成。PI3K-Akt通路和Ras/Raf/MEK/ERK通路是两个最重要的由HER受体激活的下游信号通路。来自HER受体的信号,会因为激活的HER受体在内吞作用下被内化而终止。内化后的受体要么循环回质膜(HER2、HER3、HER4),要么在溶酶体(HER1)中降解。
目前大多数关于HER2的研究都是针对于乳腺癌,这主要是因为有15%-30%的乳腺癌中有HER2的过表达。但随着对HER2的研究加深,大家现在已经认识到HER2过表达也会发生在其他形式的癌症中。因此使用表达HER2特异性嵌合抗原受体的细胞治疗也引起了大家密切关注。根据Budi等人的统计,进入临床阶段的HER2CAR-T细胞治疗项目已经有23项,其中绝大部分都在一期临床阶段。
目前细胞治疗在实体瘤领域的突破有限,因此想在HER2靶点上做出好的效果,可能还需要等待一段时间。而针对于HER2靶点的抗体药物则发展得非常成熟,例如著名的曲妥珠单抗、帕妥珠单抗以及ADC药物Kadcyla与Enhertu等等。
4、CD20
CD20是一种在正常和恶性B淋巴细胞表面表达的33-37kDa大小的非糖基化蛋白,属于MS4A(四重跨膜家族A)蛋白家族。迄今为止,除了MS4A1(编码CD20)之外,已经鉴定了18个其它的MS4A家族成员,例如高亲和力免疫球蛋白E受体β亚基(MS4A2/FcεRIβ)或HtM4基因(MS4A3)等等。MS4A蛋白是跨膜分子,预计它们具有相似的多肽序列和整体拓扑结构。
CD20蛋白由四个疏水性跨膜结构域、一个胞内结构域和两个胞外结构域(大环和小环)组成,其中N端和C端均位于胞质溶胶内。不同磷酸化会产生三种CD20的同种型(33、35和37kDa),增殖的恶性B细胞中的CD20磷酸化水平要高于静息B细胞。
通常,CD20不形成异源寡聚体,但作为同源二聚体和同源四聚体的寡聚体会存在于细胞表面,这样有助于CD20与信号转导的其他细胞表面、细胞质内蛋白相关联。有实验表明,CD20与其他四重跨膜分子(如CD53、CD81和CD82)非常接近,可以形成超分子复合物。
CD20还已知与II类主要组织相容性复合物体(MHCII)、CD40分子、BCR和与Src激酶(如LYN、FYN和LCK)相互作用的Src激酶结合蛋白(CBP)C 端有物理偶联。除了跨膜形式的CD20以外,CLL患者血浆中还存在有力的CD20,但这很可能是更大的蛋白质复合物的一部分或来源于细胞分解的细胞膜片段。
CD20在B细胞中的生物学功能及其配体仍未可知,但目前的结果可以推论出CD20直接参与了B细胞中有效BCR信号的传导。但CD20是否作为钙通道和/或其他功能来参与了这一过程,B细胞表面CD20的水平是否会影响B、T细胞相互作用或影响其他的分子途径,目前尚不知晓。
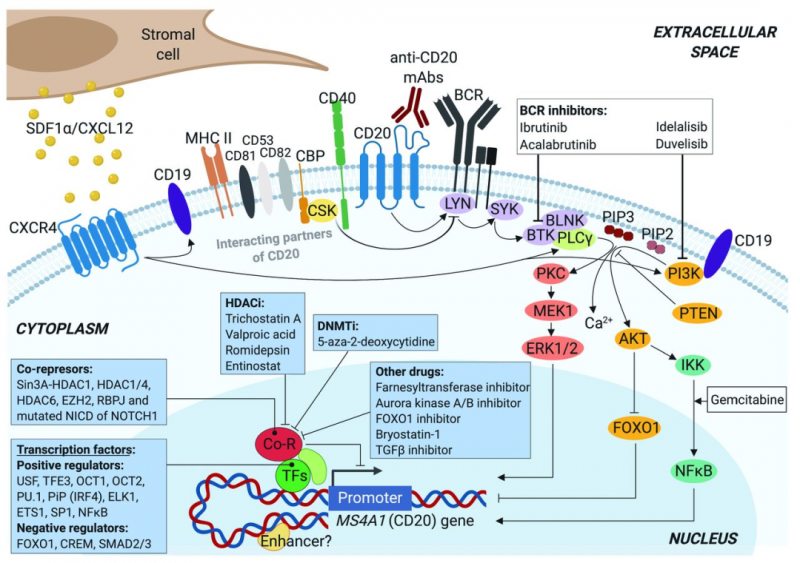
基于CD20靶点的抗体药物较多,早期的药物例如有罗氏的利妥昔单抗Rituximab,第二代的药物则往往进行了人源化或者完全人源,例如Ofatumumab、Ocrelizumab和Veltuzumab等等。
5、VEGF
血管生成是从预先存在的脉管系统生长出新血管的过程。它在胚胎发育、组织正常生长、伤口愈合、女性生殖周期以及许多疾病中起着至关重要的作用。尤其是在癌症中,如果不形成新的血液供应,肿瘤的大小就不能超过几毫米。因此血管生成对于肿瘤细胞的转移、扩散和生长是必需的。
血管内皮最重要的生长和存活因子之一,就是血管内皮生长因子(VEGF)。VEGF能够诱导血管生成和内皮细胞增殖,在调节血管生成中起了重要的作用。VEGF是一种肝素结合糖蛋白,以45kDa 的同源二聚体形式分泌。除内皮细胞外的大多数类型的细胞,都分泌有VEGF。由于最初发现的VEGF,即VEGF-A,可增加血管通透性,故被称为血管通透性因子。
此外,VEGF还可以通过刺激内皮细胞中的一氧化氮合酶,引起血管舒张,并且VEGF还可以刺激细胞迁移并抑制细胞凋亡。VEGF-A有几种剪接变体,大小分别为121、165、189和206个氨基酸。已经克隆了的VEGF家族的其他几个成员,包括VEGF-B、-C和-D。有研究显示,胎盘生长因子(PIGF)也与VEGF-A密切相关。VEGF-A、-B、-C、-D和PIGF都与platelet-derivedgrowth factors-A 与-B相关。但目前大家对VEGF-B、-C和-D的功能和调节知之甚少,但它们似乎不受调节VEGF-A的主要途径的调节。
VEGF-A的转录会因a.缺氧和b.活化的癌基因的影响而增强。
a.转录因子-缺氧诱导因子-1α(HIF-1α,在常氧中被蛋白酶体降解并在缺氧中保持稳定表达。该途径依赖于VonHippel-Lindau 基因产物)、HIF-2 α 与细胞核中的芳烃核转运蛋白(arylhydrocarbon nuclear translocator)异二聚化并与VEGF启动子/增强子结合。这是VEGF在大多数类型细胞中表达的关键途径。缺氧诱导也是VEGF-A区别于VEGF家族的其他成员和其他血管生成因子的特点之一。
b.正常氧环境中的VEGF转录则会被许多癌基因所激活,包括H-Ras和几种跨膜酪氨酸激酶,如表皮生长因子受体和ErbB2等。与正常组织相比,这些途径共同解释了肿瘤中VEGF-A表达水平显着上调的原因,并且通常具有预后意义。VEGF受体家族中有三种受体,它们具有多个类IgG细胞外结构域和具有酪氨酸激酶活性的共同特性。VEGFR1、VEGFR2和VEGFR3的酪氨酸激酶结构域被一个插入的序列所分开。
此外,内皮细胞还表达了额外的VEGF受体:Neuropilin-1和Neuropilin-2。VEGF-A可以与VEGFR1 、VEGFR2 、Neuropilin-1以及Neuropilin-2结合。PIGF和VEGF-B可以与VEGFR1 以及Neuropilin-1结合。VEGF-C和-D可以与VEGFR2 以及VEGFR3结合。
在功能上,VEGF可以通过刺激内皮细胞的增殖和存活以及通过增加血管的通透性并可以从骨髓募集血管前体细胞来促进肿瘤血管生成。与正常条件下成熟血管的形成不同,肿瘤内血管复杂、不规则且渗漏,会导致肿瘤内产生缺氧环境且造成抗肿瘤药物向肿瘤微环境的输送效率低下。
此外,VEGF对癌细胞也有一些直接作用。VEGF可能通过激活VEGFR1信号通路促进癌细胞增殖。除了在促进肿瘤血管生长中的关键作用外,VEGF还具有免疫抑制作用。VEGF可以抑制T细胞的功能,增加调节性T细胞(Tregs)和髓源性抑制细胞(MDSCs)的募集,阻碍树突状细胞(DCs)的分化和活化。
目前已经上市的针对VEGF靶点的抗体药物包括罗氏用于治疗转移性结直肠癌、转移性或复发性非鳞状细胞非小细胞肺癌的贝伐珠单抗(与可溶性VEGF结合)、礼来的治疗晚期或转移性胃癌、转移性非小细胞肺癌和转移性结直肠癌的雷莫芦单抗(Ramucirumab,VEGFR2拮抗剂,以高亲和力结合VEGFR2的细胞外结构域并阻断天然VEGFR配体:VEGF-A、VEGF-C和VEGF-D的结合)以及Genentech用于治疗视网膜静脉阻塞、湿性AMD和糖尿病性黄斑水肿后的黄斑水肿的雷珠单抗(Ranibizumab,抗VEGF-A)。目前也有针对于VEGFR2的CAR-T药物,但都还处于早期的临床研究阶段。
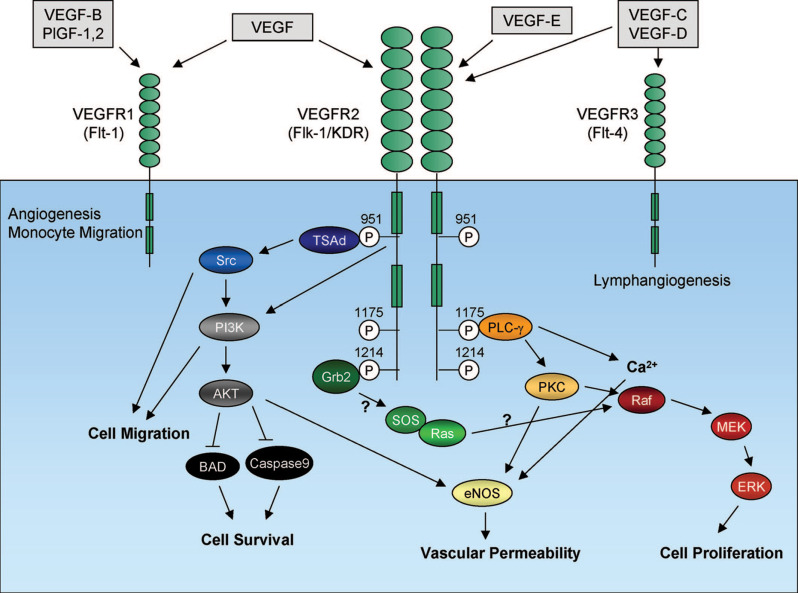
6、TNF-α
肿瘤坏死因子α(TNF-α)是一种对多种细胞类型具有多种功效的细胞因子。它已被确定为炎症反应的主要调节剂,并且参与了许多炎症反应和自身免疫疾病。
在结构上,TNF-α是一种由157个氨基酸组成的同源三聚体蛋白,主要由活化的巨噬细胞、T淋巴细胞和自然杀伤细胞产生。
在功能上,TNF-α已知会触发一系列不同的炎症分子,包括其他的细胞因子和趋化因子。TNF-α以可溶性和跨膜形式存在。
跨膜TNF-α(tmTNF-α) 是最初合成的前体形式,它可以经TNF-α转换酶(TACE,一种与膜结合的金属蛋白酶ADAMs,adisintegrin and linklloproteinase) 加工,以可溶性TNF-α的形式释放,即释放sTNF-α。加工后的sTNF-α则可以通过与1型受体(TNFR1,也被称为TNFRSF1A、CD120a和p55)和2型受体(TNFR2,也被称为TNFRSF1B、CD120b和p75)结合,产生各种生物活性。
tmTNF-α也作用于TNFR1和TNFR2,但其生物活性预计主要通过TNFR2介导。TNFR1在人体所有组织细胞中表达,是TNF-α的关键信号受体。TNFR2则通常在免疫细胞或一些特定的细胞类型,例如神经元、免疫细胞和内皮细胞中表达并促进有限的生物反应。
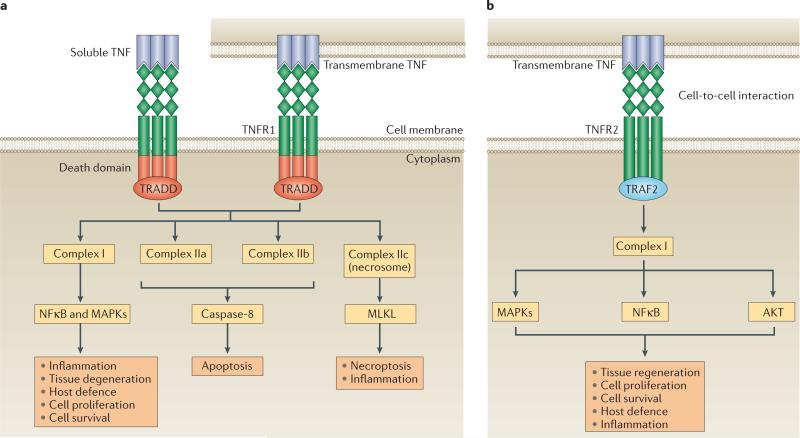
一般来说,TNF-α与其受体结合,主要是TNFR1和TNFR2,然后传递分子信号至细胞内部,以实现诱发炎症和细胞死亡等生物学功能。TNFR1由sTNF-α和tmTNF-α激活,它含有一个死亡结构域,该结构域可招募衔接蛋白TNFR1相关死亡结构域蛋白(TRADD)。
可溶性TNF或跨膜TNF连接TNFR1导致复合物I的组装,从而激活NFκB和MAPKs。TNFR1-复合物I引发的信号传导可以诱导炎症、组织退化、细胞存活和增殖,并协调针对病原体的免疫防御。与程序性细胞死亡相关的替代信号也可以在TNFR1的下游被激活。TNFR1-复合物IIa和IIb的形成可以导致细胞凋亡,而复合物IIc可以诱导坏死性凋亡和炎症。
TNFR2通过其TRAF结构域募集TNFR相关因子2(TRAF2),触发TNFR2-复合物I的形成以及下游NFκB、MAPKs和AKT的激活。TNFR2缺乏死亡结构域,因此不能直接诱导程序性细胞死亡,它主要介导稳态生物活性,包括组织再生、细胞增殖和细胞存活。该途径还可以启动炎症作用和宿主对病原体的防御。
TNF-α对病原体的防御、淋巴器官结构和生发中心形成、肉芽肿的发展、炎症的消退和组织修复来说是必需的。TNF-α能够诱导炎症、激活血管内皮、协调免疫细胞的组织募集和促进组织的破坏。失控的TNF表达,则会与炎症性疾病的发展有关,例如类风湿关节炎、炎症性肠病、银屑病、强直性脊柱炎等等。TNF在炎症(炎性疼痛)和神经元损伤(神经性疼痛)的情况下,也被观察到与痛觉过敏有关。
目前针对与TNF-α靶点的药物,主要是以中和游离的TNF-α为主要目的。其中最著名的药物,就是艾伯维的阿达木单抗(修美乐),从2012年开始,该药物连续7年都是全球销售额最高的药品。阿达木单抗是一种全人源的IgG1单克隆抗体,能够特异性阻断人TNF-α与受体的结合,因为其功能和结构与天然人IgG1相同。
阿达木单抗半衰期相对较长,约为10至13天,因此仅需要较少的皮下给药频率。其他以TNF-α为靶点的药物还包括安进的英夫利昔单抗(Remicade)、赛妥珠单抗(Cimzia)和杨森的戈利木单抗(Simponi)等等。这些药物所囊括的适应症,主要以自身免疫病为主。因为TNF-α不是一个肿瘤标志物,所以并不是细胞治疗感兴趣的靶点。
7、EGFR
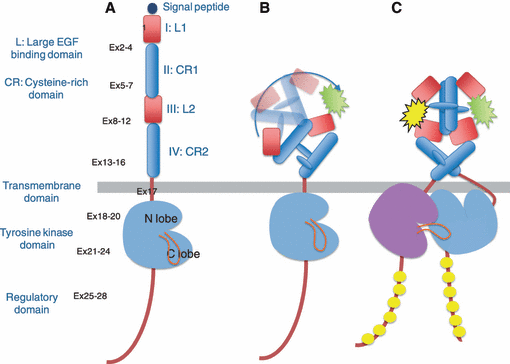
人类的肿瘤细胞会高水平表达生长因子及其受体,许多类型的恶性细胞也有表现出自分泌或旁分泌刺激的生长模式。目前研究的最为透彻的生长因子受体系统之一是EGF受体家族(也称为I型受体酪氨酸激酶或ErbB酪氨酸激酶受体)。
该家族由四种同源受体组成:表皮生长因子受体(ErbB1/EGFr/HER1)、ErbB2(HER2/neu)、ErbB3(HER3) 和ErbB4(HER4)。这些受体由一个胞外结合结构域、一个跨膜亲脂片段和一个胞内蛋白酪氨酸激酶结构域组成。前文提到的是ErbB2(HER2/neu),在这一小节会多介绍一下ErbB1/EGFr/HER1相关的背景知识。
EGFR基因位于染色体7p12-13并编码了170kDa受体酪氨酸激酶。所有ERBB蛋白都有四个功能结构域:
细胞外配体结合结构域;跨膜结构域;细胞内酪氨酸激酶结构域;和一个C端调控域。胞外结构域进一步细分为四个结构域。
酪氨酸激酶结构域由一个N-lobe和一个C-lobe组成,ATP与这两个lobe之间形成的裂缝结合。
C-末端调节结构域具有几个酪氨酸残基,它们在配体结合后被特异性磷酸化。
EGFR会在多种肿瘤中表达,包括肺、头颈、结肠、胰腺、乳腺、卵巢、膀胱和肾以及神经胶质瘤中的肿瘤。许多人类癌症中研究了EGFR表达和预后。尽管已经报道了一些差异,但具有高表达EGFR的肿瘤患者通常预后较差。
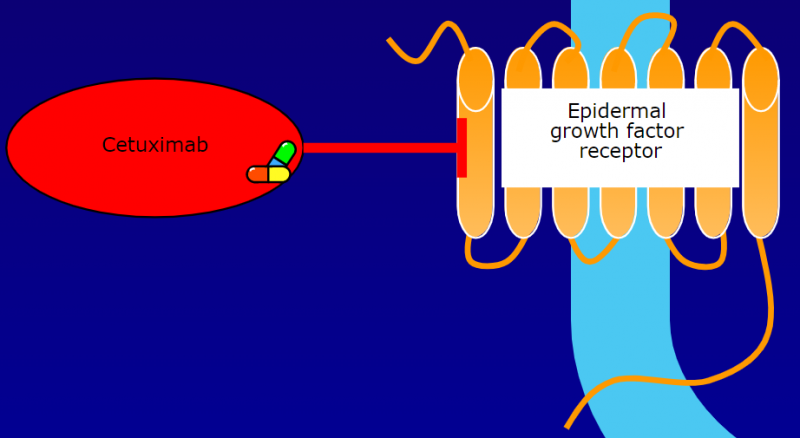
目前针对EGFR的抗体药物有很多已经上市,例如西妥昔单抗。西妥昔单抗Cetuximab(Erbitux) 是mClone Systems和百时美施贵宝的专利药,它是一种重组嵌合人/小鼠IgG1单克隆抗体,可以与正常和肿瘤细胞上的EGFR特异性结合,从而竞争性抑制正常和肿瘤组织上皮细胞产生的表皮生长因子(EGF)与其他配体的结合。
在与EGFR的结构域III(EGFR中生长因子配体的结合位点)结合后,西妥昔单抗可以抑制EGFR的构象变化,从而抑制EGFR活化,以及EGFR相关激酶(MAPK、PI3K/Akt,Jak/Stat) 的磷酸化和活化。EGFR信号通路的抑制最终可以导致细胞周期进程、细胞存活通路以及肿瘤细胞运动和侵袭的抑制。
西妥昔单抗还可以诱导细胞凋亡并减少基质金属蛋白酶和血管内皮生长因子(VEGF)的产生。在体外,西妥昔单抗被证明可以抑制肿瘤血管生成。此外,西妥昔单抗与EGFR的结合也会导致EGFR抗体-受体复合物的内化,从而导致EGFR表达的整体下调。
西妥昔单抗于2004年2月获得FDA批准,用于治疗头颈癌和转移性、K-ras野生型结直肠癌和具有BRAFV600E 突变的转移性结直肠癌(K-ras是EGFR下游的一种小G蛋白,它在促进EGFR信号级联中发挥重要作用:在一些恶性细胞中,K-ras在外显子2中发生了突变,因此无论EGFR在上游如何调节,突变的K-ras都保持激活状态。
由于K-ras的突变使其下游途径不受EGFR控制,因此西妥昔单抗对K-Ras突变的肿瘤无法发挥作用)。除此之外,西妥昔单抗还在晚期结直肠癌、表达EGFR的非小细胞肺癌(NSCLC)和不可切除的鳞状细胞皮肤癌中进行了研究。西妥昔单抗通过静脉输注给药,可作为单一疗法或与其他化学疗法联合使用,包括铂类药物、放射疗法、亚叶酸、氟尿嘧啶和伊立替康。
其他的药物例如Necitumumab是礼来的一款重组IgG1单抗,目前已被批准与顺铂和吉西他滨联合,用于转移性鳞状非小细胞肺癌(NSCLC)的一线治疗;Zalutumumab是全人源的IgG1单抗,可以用于治疗鳞状细胞癌和头颈癌;Amivantamab则是一款EGFR-MET双特异性抗体,可以用于治疗具有EGFR20 外显子插入突变的非小细胞肺癌(间充质-上皮转换因子MET)是一种在上皮细胞上表达的具有酪氨酸激酶活性的受体,其在发出信号后二聚化并激活下游的细胞分裂信号通路。
具有EGFR外显子20插入突变的NSCLC患者会对酪氨酸激酶抑制剂没有反应,并且该突变也会导致激活EGFR的构象变化。Amivantamab可以同时靶向EGFR和MET,阻止配体与受体结合,阻断信号传导,标记癌细胞,使其对自然杀伤细胞产生抗体依赖性细胞毒性,并允许巨噬细胞进行吞噬作用)。
8、间皮素
间皮素(MSLN)是一种在各种恶性肿瘤细胞上广泛过度表达的肿瘤相关抗原(包括肺腺癌、卵巢癌,以及血液系统恶性肿瘤,包括急性髓性白血病等),而其本底表达通常仅限于正常的间皮细胞,因此它是一种有很有潜力的靶向治疗target。
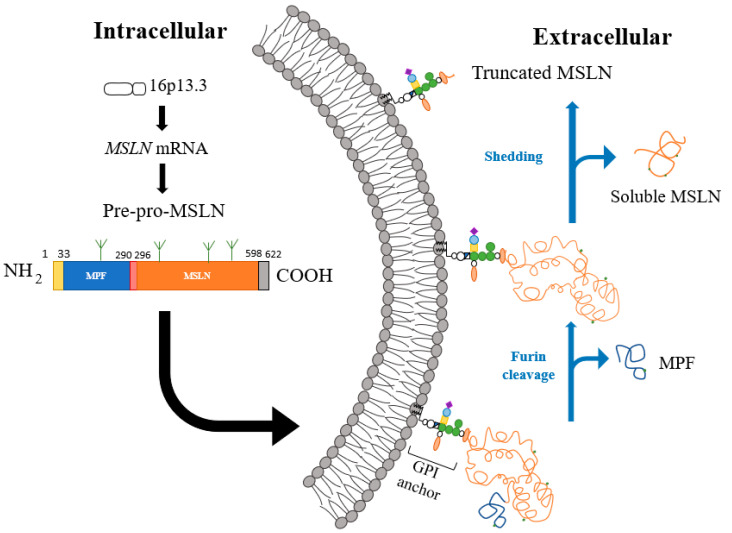
间皮素是一种与糖基磷脂酰肌醇相锚定的细胞表面蛋白,位于16p13.3的间皮素基因MSLN包含了17个外显子。2138bp的MSLN cDNA 上有一个1884bp的开放阅读框,编码一个622个AA(69kDa)的前体蛋白(pre-pro mesothelin)。
N端的信号肽由1-22位的氨基酸组成,当间皮素蛋白插入膜后会被切除。GPI锚定蛋白是指会通过C端共价结合糖磷脂酰肌醇(glycolphosphatidylinositol,GPI)而锚定在膜上的蛋白质。这些膜蛋白本身缺乏跨膜区和伸进胞质的尾部,并且都位于质膜外侧。在极性上皮细胞中它们往往靶向质膜的顶区。
因此对于间皮素来说,C-末端残基包含了599-622这一段,并在S598处添加GPI锚。间皮素的前体蛋白pre-promesothelin会在R295处的弗林蛋白酶水解位点RPRFRR处被切割,进而释放出31kDa 的分泌片段(有时称为巨核细胞增强因子;MPF),并在细胞膜上留下GPI锚定的303个氨基酸残基(40kDa) 间皮素片段(mesothelin,MSLN)。与许多GPI锚定蛋白一样,间皮素也会脱落以产生可溶性间皮素相关肽(SMRP)。
MPF和MSLN都具有生物活性,但它们的确切功能仍不清楚。最初有报道称MPF在IL-3的协助下可以刺激小鼠巨核细胞集落形成,而其在人类中的活性尚不清楚。MSLN最初被描述为在间皮瘤、卵巢癌细胞、正常间皮细胞上表达的膜蛋白。
先前的一项研究表明,MSLN似乎是正常细胞中的非必需成分,因为MSLN敲除小鼠没有出现发育或繁殖上的异常。相比之下,临床前和临床研究表明,肿瘤细胞上的MSLN异常表达在促进肿瘤细胞增殖和侵袭方面起着重要作用。MSLN也被确定为介导细胞粘附的CA125蛋白的受体。
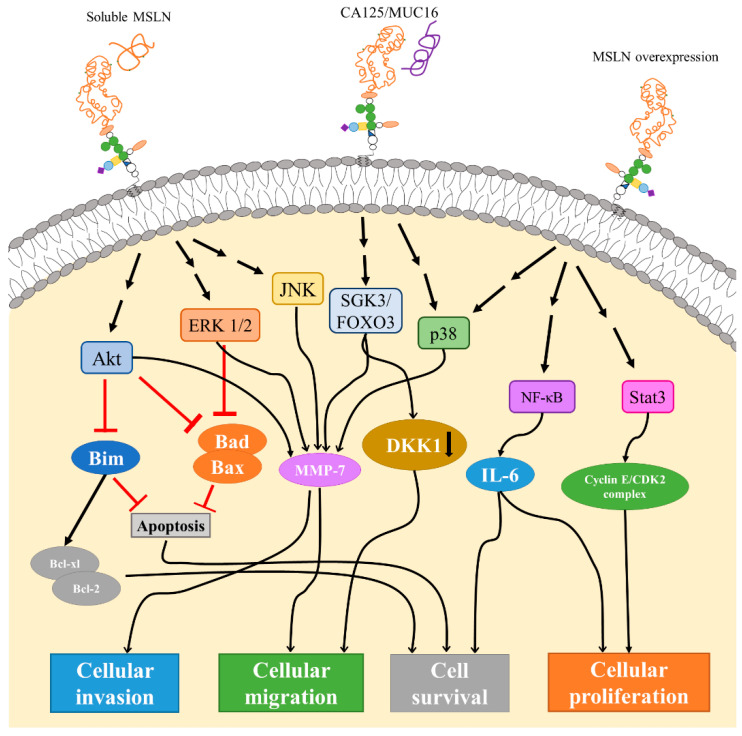
CA125和MSLN的相互作用在卵巢癌细胞腹膜植入、增加胰腺癌细胞的运动和侵袭中起着重要作用。MSLN的过表达可以激活NFκB、MAPK和PI3K通路,进而诱导细胞抗凋亡或通过诱导MMP7和MMP9的激活和表达,来促进细胞增殖、迁移和转移。根据临床试验观察,肿瘤负荷增加和总生存期差与MSLN表达的升高有关。
通过与可溶性或细胞表面间皮素的相互作用,间皮素可以触发Akt、ERK1/2和JNK信号通路。Akt和ERK1/2信号传导的下游效应包括抑制Bim、Bad和Bax,然后抑制细胞凋亡和/或刺激Bcl-xl/Bcl-2导致癌细胞存活。Akt、ERK1/2和JNK通路显示增加了基质金属蛋白酶7(MMP-7) 的表达,从而导致细胞迁移和侵袭率增加。
MMP-7通路也可以通过SGK3/FOXO3和p38通路触发。SGK3/FOXO3信号也被证明会导致DKK1的下调和表达,从而增加细胞迁移。通常,间皮素的过表达也会导致p38信号传导以及NF-kB和STAT3信号传导激活。
目前尚未有针对于间皮素的免疫治疗药物上市,但有些药品目前正在积极地推进临床研究。Amatuximab(MORAb-009) 是一种靶向间皮素的嵌合单克隆抗体,由与人IgG1和κ恒定区融合的SS1scFv 组成。Amatuximab与肿瘤细胞表面表达的MSLN结合,从而破坏细胞粘附并引发抗体依赖性细胞毒性。
抗肿瘤作用和最大耐受剂量已在I期和II期临床试验中与化疗联合治疗中确定。体外研究表明,Amatuximab抑制间皮素与CA125/MUC16的相互作用,降低了癌症转移和侵入其他组织的能力。此外,经过Amatuximab治疗后,胰腺癌细胞中的癌症干细胞标志物,如CD44、c-MET和ALDH1的表达会被下调。
Amatuximab的治疗增加了受试病人对吉西他滨的敏感性,降低了病人肝脏中c-Met和AKT的表达,同时也降低了胰腺癌细胞转移率。Anetumabravtansine,也称为BAY94-9343,是一种与DM4融合的人抗MSLN抗体ADC药物(DM4是一种主要影响增殖细胞的美登素类微管蛋白抑制剂)。
BAY94-9343与MSLN的高亲和力特异性结合可诱导有效的抗原内化。BAY94–9343在异种肿瘤模型中显示出剂量依赖性抗肿瘤功效和旁观者效应。HPN536是最新的MSLN靶向抗体药物,目前正在进行临床试验。
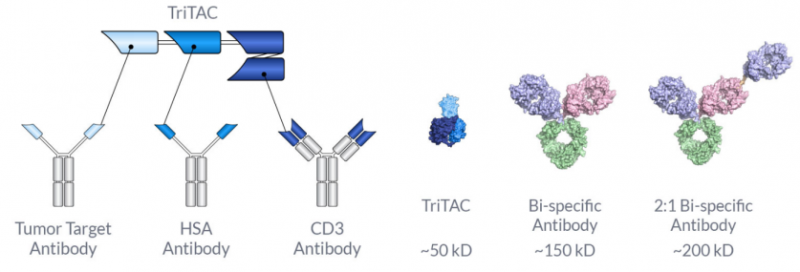
它是一种针对MSLN的TriTAC,包括三个结构域:
1.与MSLN阳性细胞结合的抗MSLN结构域;
2.延长半衰期的抗人血白蛋白结构域抗体;
3.一种与T细胞结合的抗CD3εscFv 。HPN536在MSLN存在时激活T细胞并指导T细胞杀死表达MSLN的细胞。
它的半衰期约为5天,并且在接受10mg/kg 剂量单次治疗的食蟹猴中具有良好的耐受性。HPN536的1/2a期试验(NCT03872206)是一项多中心、开放标签研究,旨在评估HPN536在多达80名与MSLN表达相关的晚期癌症患者中的安全性、耐受性、PK和活性。
9、Trop2
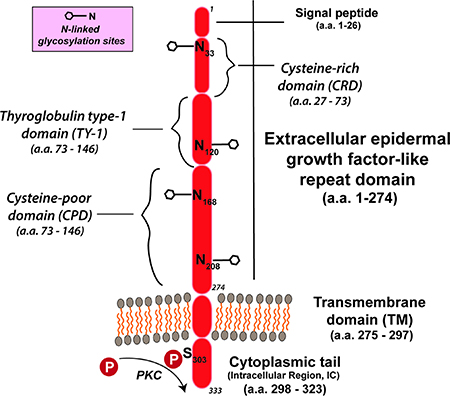
Trop2是一个约 46KDa 大的糖蛋白,在最初发现时,Trop2被描述为人类滋养层细胞的表面标志物。Trop2由 323个氨基酸组成,其中包含一个248个氨基酸的胞外结构域、一个疏水性跨膜结构域和一个短的胞质内结构域。
Trop2 的胞外结构域由GA733 1 型(Trop2synonym)motif和甲状腺球蛋白1A 型motif组成,这两种motif也存在于Trop2 的同源物Trop-1/EpCAM(上皮细胞粘附分子)中。无内含子的TACSTD2(Trop2基因)位于染色体1p32 上。
35kDa 未糖基化的Trop2 多肽在翻译后可以进行N-连接糖基化。Trop2的胞质内结构域长度为26 个氨基酸,其内包含一个磷脂酰肌醇4,5-二磷酸结合位点(PIP2 ) 并且可以被蛋白激酶C (PKC) 在其303位的丝氨酸处磷酸化。
这个磷酸化位点使 Trop2能够充当 Ca2+的信号传感器。Trop2诱导的 Ca2+递送与通过丝裂原活化蛋白激酶信号级联(MAPK) 进行的细胞周期有关。Trop-2的表达可以由几种促癌转录因子(例如,CREB1、NF-κB和 HOXA10等)通过正反馈调节。
Trop-2表达也可能由于几种转录因子(例如,HNF4A、TP63/TP53L、ERG、HNF1A/TCF-1和 FOXP3)的失活而上调。Trop-2的过表达会加速癌细胞周期并推动癌症生长。
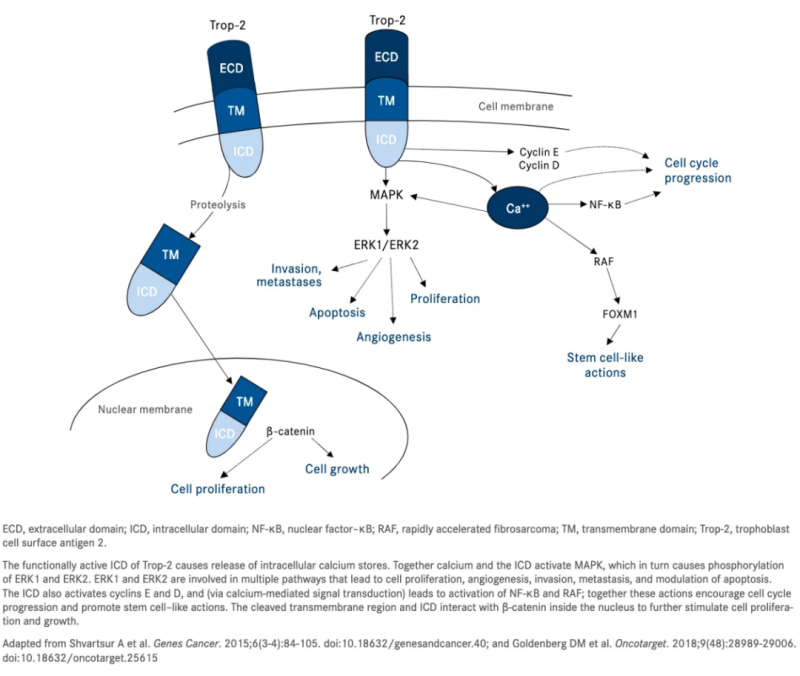
Trop-2最初被认为只是细胞内钙信号的传感器。然而,现在已知它可以在与肿瘤相关的多种信号通路中发挥重要作用。Trop-2作为钙信号传感器在细胞内表达,可以导致钙从内部储存中被调动。增加的细胞内钙水平激活MAPK,进而提高磷酸化ERK1 和ERK2 的水平。
ERK1和 ERK2是调控细胞周期进程、血管生成、细胞增殖、细胞侵袭和转移的重要介质。细胞内的钙还可以激活可刺激细胞生长的NF-κB通路和对FOXM1(人类实体瘤中最常见的过表达基因之一)上调至关重要的RAF 通路。
Trop2可以与多种配体结合,从而发挥功能,例如IGF-1、claudin-1和 cyclin D1 和PKC。IFG-1(胰岛素样生长因子1,也称为生长素C,是一种分子结构与胰岛素相似的激素,在儿童成长中起重要作用,对成人的合成代谢也有作用。
IGF-1主要由肝脏产生,并受到生长激素(GH)调控。大部分IGF-1 与6 种结合蛋白(IGF-BP) 中的一种结合。IGFBP-1的表达则受胰岛素调节。)是Trop2的配体,它可以使Trop2能够激活下游介质(PIP2和 Ca2+)并调节 IGF-1R信号传导。
Trop2的 PIP2结合结构域可以结合IGF-1,因此在竞争中胜过IGF-1 结合蛋白。Trop2功能的另一种可能性是它可能与IGF-1 形成复合物,从而阻止IGF-1R的信号传导。跨膜蛋白claudin-1 和claudin-7 是Trop2 胞外域(EC)的结合配体。它们在上皮屏障的紧密连接中起重要作用。
科研人员推测Trop2 在claudin重排过程中可能会充当锚点或转运蛋白,又或者它可能充当稳定剂以防止claudin 被泛素-蛋白酶体系统所降解。此外,Trop2在保持细胞间紧密连接完整性方面也很重要。
Trop2可能间接影响蛋白质之间的相互粘附作用,因为它可以通过P1 整合素/RACK1(活化蛋白激酶C的受体)复合物的形成来调节细胞对纤连蛋白的粘附。Trop2的缺失导致某些蛋白质亚细胞定位的表达和重排降低,从而影响上皮屏障的性能。
由于 Trop-2是几种实体瘤类型中临床相关的细胞表面抗原,它在癌细胞上的过表达使其成为特定疗法靶向的理想候选者。目前,FDA已经批准了一款Trop-2 靶向的ADC 药物Sacituzumab govitecan-hziy,其他几款药物也正处于临床前和临床开发阶段。
FDA批准了Sacituzumab govitecan-hziy用于治疗转移性TNBC,并且还给予了Sacituzumab govitecan-hziy治疗转移性尿路上皮癌、非小细胞肺癌和小细胞肺癌的快速通道指定。Sacituzumab govitecan-hziy 的成分已经过优化,可有效靶向表达Trop-2 的肿瘤。
在Sacituzumab govitecan-hziy中,人源化单克隆抗体(hRS7) 与Trop-2 结合并将govitecan (SN-38)递送至细胞表面。SN-38是伊立替康的活性代谢物,是DNA拓扑异构酶 I抑制剂。可水解的CL2a 接头将SN-38 与hRS 7 共价结合。当在细胞内释放时,SN-38会导致双链 DNA断裂,从而导致细胞凋亡。
此外,可水解接头允许部分SN-38有效载荷释放到肿瘤微环境中,从而通过旁观者效应杀死相邻的肿瘤细胞。除此之外,第一三共(DS-1062)、科伦药业(SKB264)等药企均有针对该靶点的药物走到了二期临床。
10、Claudin
Claudin密蛋白是一类跨膜蛋白,是细胞间紧密连接(Tightjunction,TJ)的重要组成部分,它们对于调节细胞通透性和维持上皮细胞极性至关重要。目前在人类细胞中,已鉴定出 24个密蛋白基因中的23个(CLDN13除外),而在小鼠和大鼠中检测到全部24个成员。
人类基因组包含成对的CLDN基因,它们具有相似的序列并且在染色体上的位置也很近,例如16号染色体上有CLDN6和CLDN9,4号染色体上有CLDN22和CLDN24,21号染色体上有CLDN8和CLDN17,以及7号染色体上有CLDN3和CLDN4。这可能表明部分Claudin基因是由另一段基因复制产生的,相邻基因甚至可能是协同调控的。
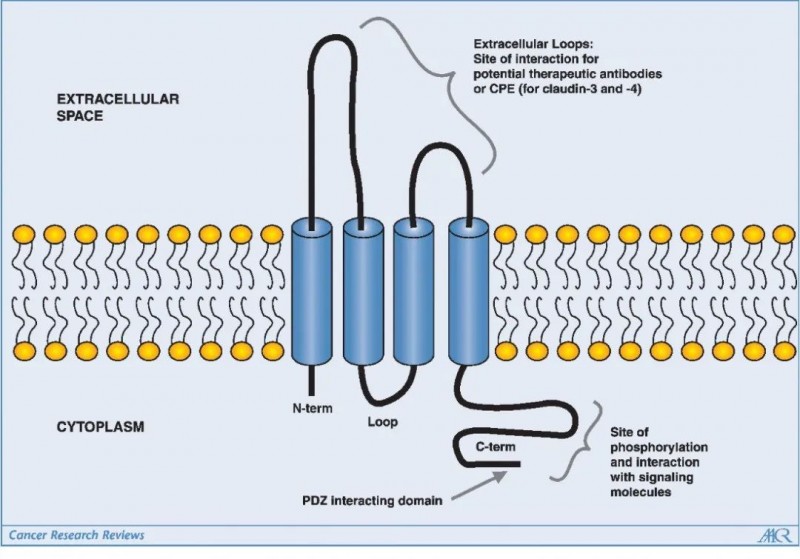
大多数密蛋白的大小在20-34kDa范围内并且有四个跨膜螺旋,其氨基和羧基末端的尾部延伸到细胞质中。此外,密蛋白有两个细胞外环,其中一个细胞外环含有带电氨基酸,在细胞旁离子选择性中起关键作用。不同Claudin蛋白羧基末端尾部的大小和序列大多不同,但通常都包含一个PDZ结构域结合基序,允许Claudin直接与细胞质TJ相关蛋白如ZO-1、ZO-2、ZO-3和MUPP1相结合。
此外,羧基末端尾部区域是翻译后修饰(例如磷酸化)的位点,这会影响Claudins的定位和功能:其中包括MAPK或蛋白激酶C(PKC) 对Claudin-1的磷酸化,以及环AMP(cAMP) 诱导的Claudin-5的磷酸化可以促进TJ的屏障功能;PKA介导的Claudin-16磷酸化可以增加Mg2+的转运;其他蛋白质,如突变型 WNK赖氨酸缺陷蛋白激酶4(WNK4) 也通过磷酸化密蛋白来增加细胞旁通透性。
科研人员在多种癌症中,都检测到了Claudin蛋白表达水平的改变,特别是Claudin-1、-3、-4和-7。某些密蛋白(包括密蛋白1和密蛋白7)在浸润性乳腺癌、前列腺癌和食管癌中下调了表达水平。而与之相反,Claudin-3和Claudin-4的上调也与肿瘤发生有关。
与正常卵巢组织相比,Claudin-3和Claudin-4在包括浆液性癌在内的卵巢癌中高度过表达,并且它们在其他几种恶性肿瘤中的表达水平也有上调,其中包括乳腺癌、胃癌、胰腺癌、前列腺癌和子宫癌。
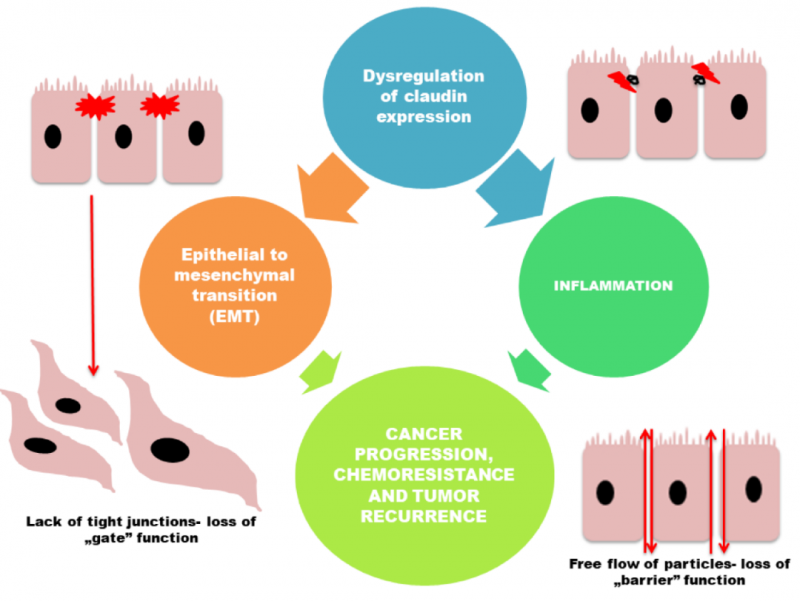
因此,可能在不同癌症中,Claudin的表达变化发挥着不同的作用。例如上皮肿瘤细胞TJ失去功能,导致在肿瘤发生过程中细胞极性丧失和上皮完整性受损。因此,Claudin表达的缺失,可能会影响与细胞粘附缺失相关联的肿瘤进展。
然而,也有多项研究表明,密蛋白表达的增加也可能通过帮助细胞迁移、侵袭和转移,促进肿瘤进展。除了这种表达异常之外,Claudin蛋白的错误定位也有可能助于它们在肿瘤发生中的作用。例如,膀胱肿瘤中Claudin-1和Claudin-4以及Claudin-7定位改变可以对食管癌的侵袭性产生影响。
此外,Claudins的功能受翻译后修饰(如磷酸化)的调节。PKA或PKC磷酸化密蛋白羧基末端结构域中的丝氨酸和/或苏氨酸位点会影响它们在癌细胞中的作用,如在卵巢癌细胞中,它会增加细胞旁通透性。
目前有很多药厂都有布局针对Claudin靶点的药物,尤其是Claudin18.2,目前大量的本土药企扎堆于该靶点,例如君实(JS-012)、信达(IBI-389)、再鼎(ZL-1211)、荣昌生物(RC-118)、石药(SYSA-1801)、安进(Gresonitamab)都有药物进入临床研究阶段,CAR-T药物中,南京传奇(LB-1908、LB-1904)、科济生物(CLDN18.2CAR-T)、信达生物(IBI-345)等公司也都有针对该靶点的管线。
11、CTLA-4与PD-1、PD-L1
T细胞的生命始于胸腺。未成熟的T细胞在胸腺中增殖并通过TCR基因片段的重组产生广泛的TCR库。然后开始选择过程,对自身肽具有强烈反应性的T细胞在胸腺中被剔除,以防止在中枢耐受的过程中发生自身免疫反应。与MHC结合能力不足的T细胞会发生凋亡,但对MHC分子和自身肽反应较弱的T细胞不会被清除,而是作为幼稚细胞释放,在血液、脾脏和淋巴器官中循环。
在那里,它们将暴露于展示了外来抗原(在机体被感染的情况下)或突变的自身蛋白(在机体存在恶性肿瘤的情况下)的专业APC。一些TCR可能具有与自身抗原发生交叉反应的特性。为了防止自身免疫,许多免疫检查点通路可以在免疫反应期间的多个步骤中调节T细胞的激活,这一过程被称为外周耐受。
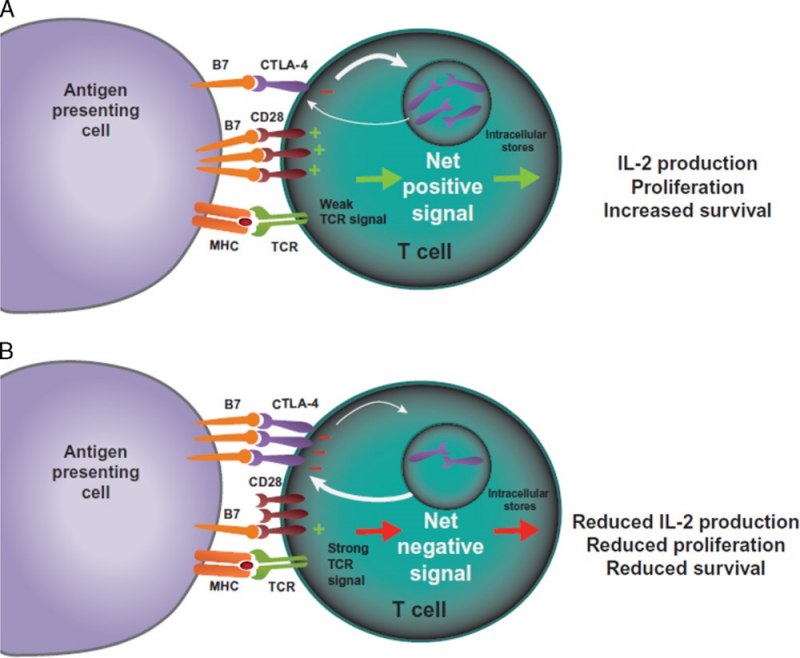
外周耐受过程的核心是细胞毒性T淋巴细胞相关抗原4(CTLA-4) 和程序性死亡1(PD-1)两个免疫检查点途径。CTLA-4和PD-1通路被认为会在免疫反应的不同阶段发挥作用。CTLA-4被认为是免疫检查点抑制剂的“领导者”,因为它在初始 T细胞激活的初始阶段(通常在淋巴结中)就可以阻止潜在的自身反应性T细胞的激活。
PD-1通路则是负责在免疫反应的后期,调节先前已经被激活的T细胞,并且主要是在外周组织中。T细胞活化是一个复杂的过程,需要多于1个刺激信号。TCR与MHC结合提供了对T细胞激活的特异性,但同时还需要一个共刺激信号。APC上的B7-1(CD80) 或B7-2(CD86) 分子可以与T细胞上的CD28分子结合,进而导致T细胞内的信号传导。
足够水平的CD28-B7结合可以提高T细胞增殖水平、增加T细胞存活率,并可以通过产生细胞生长因子如白细胞介素2(IL-2)来增加能量代谢和上调细胞生存基因的表达。
CTLA-4是一种CD28的同源物,对B7具有更高的结合亲和力;然而,与CD28不同的是,CTLA-4与B7的结合不会产生刺激信号。因此,这种竞争性结合可以减少通常由CD28-B7结合所提供的共刺激信号。CD28-B7结合与CTLA-4-B7结合的相对数量决定了T细胞是被激活还是毫无反应。此外,另一些证据表明CTLA-4与B7的结合实际上可能产生抑制信号,以此抵消来自CD28-B7、TCR-MHC结合的所产生的刺激信号。此类抑制信号的可能机制包括直接抑制TCR免疫突触、抑制CD28或其信号通路,或使T细胞的流动性增加从而导致使其与APC相互作用的能力下降等等。
PD-1是B7/CD28共刺激受体家族的成员。它通过与其配体:程序性死亡配体1(PD-L1) 和程序性死亡配体2(PD-L2) 的结合来调节T细胞活化。与CTLA-4的信号传导类似,PD-1与其配体结合后,可以抑制T细胞增殖、抑制干扰素-γ(IFN-γ)、肿瘤坏死因子-α和IL-2的产生,并降低T细胞的存活率。如果T细胞同时经历其TCR和PD-1与配体结合,PD-1所产生的信号会阻止关键TCR信号中间体的磷酸化,从而终止早期TCR信号传导并减少T细胞的活化。PD-1的表达是T细胞耗竭的标志,这些T细胞往往经历了高水平的刺激或CD4+T细胞帮助的减少。这种耗竭的状态往往发生在慢性感染和癌症期间,其特点是T细胞的功能障碍,从而导致对感染和肿瘤的控制不佳。
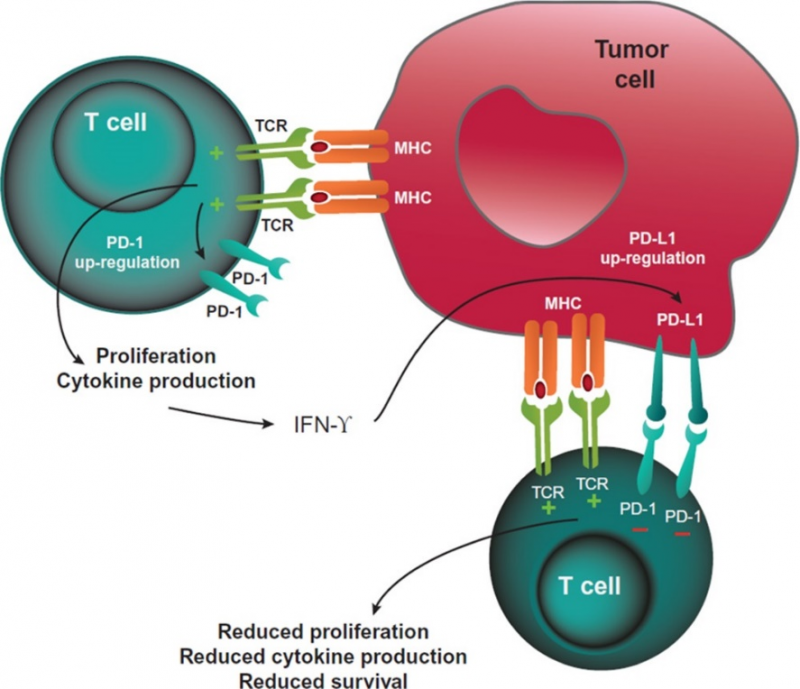
癌症免疫疗法的一个核心概念是:会被T细胞识别并杀伤的肿瘤细胞会有利用外周耐受性来逃避宿主免疫系统的方法,因此可以使用药物来恢复免疫系统对癌细胞的识别。目前已经有大量的PD-1/PD-L1的药物被批准上市且获得商业上的成功,针对抑制免疫检查点CTLA-4,目前也有一个单抗药物已被批准,即BMS的Lpilimumab,此外,阿斯利康的治疗间皮瘤、肝癌的Tremelimumab与BMS用于治疗风湿性疾病,如类风湿或银屑病关节炎的Abatacept目前也获得临床上的进展或被批准上市。
12、CD38
CD38是一种II型跨膜糖蛋白,分子量为46kDa。CD38于1980年代初由ELReinherz 和SFSchlossman 首次发现,当时被命名为T10,并作为胸腺细胞和活化T细胞的标志物来研究。CD38含有一个短的氨基末端胞质尾、一个单次跨膜区和一个长的细胞外羧基末端结构域。CD38不仅在浆细胞上高度表达,而且在其他淋巴、骨髓细胞、红细胞和血小板上也有一定的的表达水平。
有研究人员对CD38在这些细胞群中的表达水平进行了比较,结果发现,除了浆细胞之外,NK细胞表达CD38的水平最高,其次分别是B细胞和T细胞。CD38也在非造血来源的组织中表达,包括前列腺上皮细胞、胰岛细胞,以及一些神经元的外核和树突。其他CD38+细胞包括气道横纹肌细胞、肾小管、视网膜神经节细胞和角膜细胞。
在非实体瘤中,CD38被认为是B细胞慢性淋巴细胞白血病(B-CLL)的预测因子,在外周血B淋巴细胞上CD38高表达的B-CLL患者有着明显的预后不良。其他确定了CD38高表达的非实体瘤病症还包括,巨球蛋白血症、全身性轻链(AL)淀粉样变性、套细胞淋巴瘤/MCL、急性髓性白血病/AML、急性淋巴细胞白血病/ALL和NK细胞白血病。其中值得注意的是,CD38在多发性骨髓瘤(MM)细胞上的表达非常高,并且CD38可以通过其酶活性抑制机体对肿瘤细胞的免疫反应。
在一些实体瘤中,腺苷的增加与CD38的高水平表达相关,并且腺苷对机体的免疫反应具有显着的抑制作用。CD38过表达有助于在肿瘤微环境中产生腺苷。然后腺苷招募免疫抑制细胞,如Treg细胞、髓源性抑制细胞/MDSC细胞、癌症相关成纤维细胞/CAF细胞等,从而导致免疫系统紊乱。
此外,腺苷还可以与免疫细胞表面的受体A2AR结合以激活抑制性信号通路。总而言之,在肿瘤微环境中,CD38可能通过腺苷受体信号抑制CD8+T细胞等免疫反应。尽管CD38在实体瘤中的特定机制仍有待进一步探索,但一些研究已经阐明了CD38在黑色素瘤、神经胶质瘤、食道癌、宫颈癌和肺癌中似乎起到促肿瘤因子的作用。
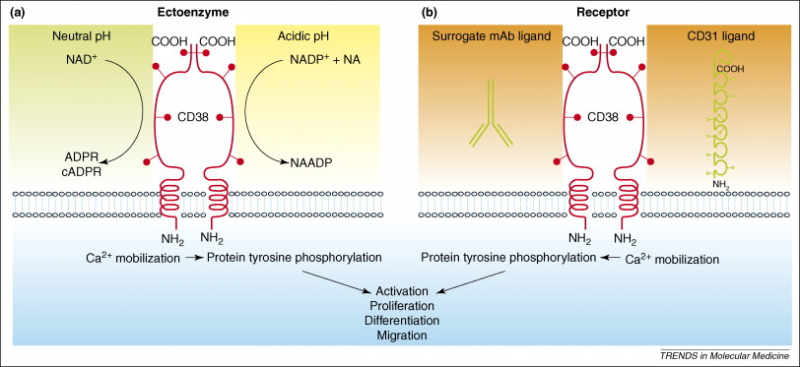
在功能上,CD38可以起到以下两种作用:
a.作为受体。科研人员在观察到激动性抗体结合CD38进而能够诱导信号后,推断出CD38可能是一种受体。这些信号依赖于辅助细胞和IL-2,并与CD3和CD2的功能相叠加。后来据报道,CD38分子参与触发细胞的激活和增殖信号。随着内皮细胞上的CD31被鉴定为自然杀伤细胞上CD38的特异性配体后,CD38目前已被普遍认为是一种受体,尽管由于它在细胞质内的结构域非常短,并且就它本身无法发挥这种功能。
b.作为胞外酶。CD38是一种多功能的胞外酶(在大多数情况下,CD38定向在膜内,但是其催化结构域位于细胞外;因此,它作为一种胞外酶,发挥作用的位置在细胞外),能够参与烟酰胺腺嘌呤二核苷酸(NAD)和烟酰胺腺嘌呤二核苷酸磷酸(NADP)的分解代谢,这是目前已经确定的两种主要分子底物。看起来CD38在胞外分解代谢NAD,而大多数NAD是在细胞内,这两点好像是自相矛盾的,但有研究证明,CD38可以在NAD前体被吸收到细胞中并进入NAD生物合成途径之前在胞外分解它们。该反应可以产生有效的细胞内钙离子动员化合物(ADP ribose和NAADP等)。此外,如前文所说,CD38还参与了腺苷的产生。
在药物方面,2015年,达雷妥尤单抗Daratumumab创造了历史,成为第一个获批用于治疗MM和轻链淀粉样变性的针对CD38的mAb药物。Daratumumab是一种IgG1kappa 单克隆抗体,由Janssen和Genmab开发,它可以靶向并诱导高表达CD38的细胞的凋亡。除此之外,Isatuximab也于2020年3月2日获得批准,用于多发性骨髓瘤的治疗。它由美国Sanofi-Aventis公司生产,是一种人源化、IgG1衍生的单克隆抗体。
除此之外,安进、Glenmark与YZYBiopharma也有针对于CD38/CD3的双抗药物进入临床阶段(AMG-424、GBR-1342、Y-150)。最近的研究结果也表明,CD38也是细胞治疗的良好靶点。CAR-T细胞疗法针对MM相关抗原,例如CS1、BCMA、SLAMF7和CD19在临床前模型和/或临床试验中被证明是有效的。因此抗CD38-CAR-T 细胞疗法也可能成为MM患者的一种新型的有效治疗工具。目前优卡迪生物、Sorrentotherapeutics等公司均在CD38-CAR-T药物上有所布局。
内容来源:细胞与基因治疗领域
























